
Отклонение поведение растворов кислот
Отклонение поведение растворов кислот, оснований и солей от законов, выведенных для разбавленных растворов. В предыдущей главе мы познакомились с теорией растворов Вант-Гоффа, объясняющей важнейшие свойства разбавленных растворов, исходя из аналогии между растворами и газами. Все выводы этой теории блестяще подтверждались результатами экспериментальных исследований, производившихся с растворами очень многих веществ. Лишь к одной категории растворов установленные, закономерности оказались неприменимыми. Это были растворы обыкновенных солей, а также растворы большинства кислот и растворимых оснований. Их осмотическое давление, вычислявшееся по понижению давления пара или понижению точек замерзания и повышению точек кипения, неизменно оказывалось значительно большим, чем требовала теория. Наоборот, молекулярные веса, определенные на основании измерения тех же величин, всегда получались меньше истинных.
Приведем пример. При растворении 1 г поваренной соли в 100 г воды температура замерзания понижается на 0,617°. Так как криоскопическая константа для воды равна 1,86°, то, вычисляя молекулярный вес поваренной соли, находим:
M = (1,86 x 10) :0,617 = 30,1
В действительности молекулярный вес поваренной соли равен 58,5, т. е. почти вдвое больше.
Если же по правильному молекулярному весу рассчитать, насколько в данном случае должна понизиться температура замерзания, то получим:
∆t = (1,86 x 10) : 58,5 = 0,318°
Таким образом, наблюдаемое понижение температуры почти вдвое больше теоретического. А так как понижение температуры замерзания пропорционально осмотическому давлению, то, следовательно, и осмотическое давление указанного раствора больше «нормального», предусматриваемого теорией Вант-Гоффа.
Такое же отклонение от теории обнаружено и для растворов других солей, а также для растворов большинства кислот и оснований.
Величина осмотического давления выражается уравнением
Р = CRT
Чтобы распространить это уравнение на растворы с «ненормальным» осмотическим давлением, Вант-Гофф ввел в него поправочный коэффициент i (называемый также изотоническим коэффициентом), показывающий, во сколько раз осмотическое давление данного раствора больше нормального: Р = iCRT
Коэффициент i определялся для каждого раствора экспериментальным путем или по понижению давления пара или по понижению точки замерзания и повышению точки кипения. Так как все эти величины пропорциональны осмотическому давлению, то, установив, во сколько раз та или иная из них больше вычисленной теоретически, тем самым узнавали, во сколько раз осмотическое давление раствора больше нормального.
Обозначим через Р’ осмотическое давление раствора, через
∆t кип — повышение точки кипения, ∆t’зам—понижение точки замерзания раствора, не подчиняющегося законам Вант-Гоффа и Рауля, а через Р, ∆t кип и ∆tзам— значения тех же величин, вычисленные теоретически по молярной концентрации раствора. Тогда коэффициент i выразится следующими отношениями:
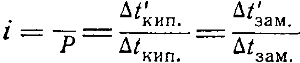
Значения коэффициента найденные Вант-Гоффом для 0,2 н. растворов некоторых солей по понижению их точек замерзания, приведены в табл. 10.
Таблица 1
| Значение коэффициента i для 0,2 н. растворов некоторых солей | ||||
| Соль | Формула | Понижение точки замерзания | ||
| вычисленное по
формулам Рауля ∆tзам. |
наблюдаемое
∆t’зам |
i = ∆t’зам : ∆tзам | ||
| Калий хлористый
Калий азотнокислый Магний хлорнокислый Кальций азотнокислый |
КСl
KNO3 MgCl2 Са (NO3)2 |
0,372
0,372 0,186 0,186 |
0,673
0,664 0,519 0,461 |
1,81
1,78 2,79 2,48 |
Данные табл. 10 показывают, что коэффициент i для различных солей различен. Кроме того, оказалось, что он растет с разбавлением раствора, приближаясь к целым числам 2, 3, 4. Для солей аналогичного состава эти числа одинаковы. Например, для всех солей, образованных одновалентными металлами и одноосновными кислотами, при достаточном разбавлении их растворов коэффициент i становится равным 2; для солей, образованных двухвалентными металлами и теми же кислотами, — равным 3, и т. д.
Итак, кислоты, основания и соли, растворяясь в воде, создают значительно большее осмотическое давление, чем эквимолекулярные количества всех остальных веществ.
Как же объяснить это явление не вступая в противоречие с теорией Вант-Гоффа?
Укажем прежде всего, что аналогичное явление наблюдается в отношении некоторых газов или веществ, находящихся в газообразном состоянии.
Например, пары хлористого аммония NH4Cl, пяти хлор истого фосфора PCl5, иода и других веществ при нагревании в закрытом сосуде обнаруживают более высокое давление, чем следует по закону Гей-Люссака. Наоборот, молекулярный вес их, вычисленный на основании определения плотности пара, оказывается ниже теоретического.
В случае газообразных веществ это явление легко объясняется диссоциацией. Если, положим, хлористый аммоний NH4Cl разложится на молекулы NH3 и НСl, то понятно, что при неизменном объеме давление, зависящее от числа частиц, должно увеличиться вдвое. При неизменном же давлении объем газа должен возрасти вдвое, а следовательно, плотность должна вдвое уменьшиться. При неполной диссоциации, когда только часть молекул подверглась разложению, давление имеет некоторую среднюю величину.
Естественно было предположить, что и в растворах, обладающих ненормально высоким осмотическим давлением, молекулы растворенного вещества распадаются на какие-то более мелкие частицы. Но так как осмотическое давление зависит не от весового количества растворенного вещества, а от числа частиц, находящихся в единице объема раствора, то с увеличением их числа оно тоже увеличивается.
Такое предположение впервые было высказано в 1887 г. шведским ученым Аррениусом и легло в основу его теории, объясняющей поведение кислот, оснований и солей в водных растворах. К этой теории Аррениус пришел путем изучения электропроводности растворов.
Вы читаете, статья на тему Поведение растворов кислот
Добавить комментарий
Для отправки комментария вам необходимо авторизоваться.